 Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY



 Indexed Articles
Indexed ArticlesSelect your language of interest to view the total content in your interested language
Guzmán-Casta Jordi1*, Alatorre-Alexander Jorge Arturo1, Carrasco-Cara Chards Sonia2, Guzmán-Huesca Jorge3, Riera-Sala Rodrigo1, Sánchez-Ríos Carla Paola1, Peña-Mirabal Erika Sagrario5, Hernández-Dehesa Itzel Ariadna6, Baltazar-Contreras Rocíodel Carmen4 , Martínez-Barrera Luis Manuel1, Rodríguez-Cid Jerónimo Rafael1
1Clinic of Thoracic Oncology, Instituto Nacional de Enfermedades Respiratorias, Dr. Ismael Cosío Villegas, Ciudad de México, México
2Facultad de Medicina, Universidad Nacional Autónoma de México, Ciudad de México, México
3Internal Medicine, Bonita Community Health Center, Florida, USA
4Resident Clinical Oncology, Hospital General de México, Dr. Eduardo Liceaga, Ciudad de México, México
5Pathology Department, Instituto Nacional de Enfermedades Respiratorias, Dr. Ismael Cosío Villegas, Ciudad de México, México
6Radiology Department, Hospital Ángeles Acoxpa, Ciudad de México, México
Correspondence to: Guzmán-Casta Jordi, Clinic of Thoracic Oncology, Instituto Nacional de Enfermedades Respiratorias, Dr. Ismael Cosío Villegas, Ciudad de México, México; E-mail: jordioncomed@gmail.com
Received date: September 21, 2020; Accepted date: October 1, 2020; Published date: October 8, 2020
Citation: J Jordi GC, Jorge Arturo AA, Chards Sonia CC, et al. (2020) Primary Pulmonary Angiosarcoma: A Case Report and Literature Review. J Med Res Surg 1(5): pp.1-4
Copyright: ©2020 Jordi GC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Primary sarcomas of the thorax are rare. They are classified according to their histologic features and constitute a large group of tumors that occur in the lung, mediastinum, pleura, and chest wall. Although primary thoracic sarcomas commonly manifest as large, heterogeneous masses, they have a wide spectrum of radiologic manifestations, including solitary pulmonary nodules, central endobronchial tumors, and intraluminal masses within the pulmonary arteries. Angiosarcomas are rare, malignant vascular tumors, representing about 2% of all soft tissue sarcomas. The most frequently primarily affected sites include the heart, liver, breast, skin, and scalp, and they have a high rate of metastases to the lungs and, less commonly, liver, regional lymph nodes, and bone. Definitive diagnosis is made based on histopathological and immunohistochemical findings. No standard treatment regimen has been established for pulmonary angiosarcoma. Radiation therapy, chemotherapy, surgical resection, and immunotherapy have been attempted. The use of radiation therapy in conjunction with surgery has resulted in local control and excellent functional and cosmetic outcome, for patients with angiosarcoma of the head and neck, but not the lung. For advanced-stage disease, other than the combination of doxorubicin and ifosfamide-based regimens, there have been few other chemotherapeutic options for the treatment of local or metastatic angiosarcoma.
Pulmonary, Angiosarcoma, Tumors, Chemotherapy
Primary sarcomas of the thorax are rare. The diagnosis is established only after sarcoma-like primary lung malignancies and metastatic disease have been excluded. Primary sarcomas of the thorax are classified according to their histologic features and constitute a large group of tumors that occur in the lung, mediastinum, pleura, and chest wall. Angiosarcoma, leiomyosarcoma, rhabdomyosarcoma, and mesothelioma (sarcomatoid variant) are the most common primary intrathoracic sarcomas. Although primary thoracic sarcomas commonly manifest as large, heterogeneous masses, they have a wide spectrum of radiologic manifestations, including solitary pulmonary nodules, central endobronchial tumors, and intraluminal masses within the pulmonary arteries [1].
At most, a few hundred new cases of angiosarcomas (100-400) are diagnosed every year in the United States. Roughly 50% of these cases occur in the head and neck, with the next most common origin being the soft tissue of the lower extremities [2].
Angiosarcomas are rare, malignant vascular tumors, representing about 2% of all soft tissue sarcomas. The most frequently primarily affected sites include the heart, liver, breast, skin, and scalp, and they have a high rate of metastases to the lungs and, less commonly, liver, regional lymph nodes, and bone. Primary extrapulmonary angiosarcomas originating in the chest include those in the heart, aorta, or great vessels and, because of their rarity, are poorly characterized [3]. They are a subtype of soft tissue sarcomas which are aggressive, malignant endothelial cell tumors of vascular or lymphatic origin [4].
Epidemiologically, there is no gender priority and the average age of occurrence is 45 years. Predisposing factors are unknown, may be related to certain stimulating factors such as acrylic, chronic empyema, and tuberculous pyothorax [5].
Early diagnosis is not common because of the rarity of angiosarcoma in the lung and hence, low index of suspicion. Chest radiography may reveal a spectrum of findings ranging from normal to multiple nodular densities with or without pleural effusions to diffuse alveolar infiltrates compatible with pulmonary haemorrhage [6].
Definitive diagnosis is made based on histopathological and immunohistochemical findings. The microscopic feature is always misleading except for some subtle clues such as red blood cells containing intracytoplasmic lumina. Immunohistochemically, the neoplastic cells showed reactivity for endothelial cell biomarkers (CD31, CD34, factor VIII, FLI‑1, and Ulex europaeus agglutinin I), epithelial biomarkers (cytokeratins and sometimes epithelial membrane antigen), and vimentin [5]
No standard treatment regimen has been established for pulmonary angiosarcoma. Radiation therapy, chemotherapy, surgical resection, and immunotherapy have been attempted. For patients with localized disease, surgery is the most effective. Surgery must be considered as early as possible if the tumor is deemed resectable. Two chemotherapy regimens that have demonstrated full and partial effects are doxorubicin/ ifosfamide and docetaxel/gemcitabine. Immunotherapy with recombinant interleukin 2 in combination with either surgery or chemotherapy has also been shown to be effective. The use of radiation therapy in conjunction with surgery has resulted in local control and excellent functional and cosmetic outcome, for patients with angiosarcoma of the head and neck, but not the lung [7].
20-year-old male with no significant past medical history other than a motor vehicle accident a year ago requiring a Thoracotomy who presents with a 4-month history of “in crescendo” left thoracic pain associated with hemoptysis and a10 kg weight loss. The patient was seen at the Emergency Room at the National Institute of Respiratory Diseases (INER) where a CT of the Chest revealed a mediastinal mass encircling the heart and great vessels measuring 8 × 10 centimeters and multiple bilateral pulmonary metastases. Additional studies like Bone Scan, CT of the abdomen and brain were negative (Figure1,2).
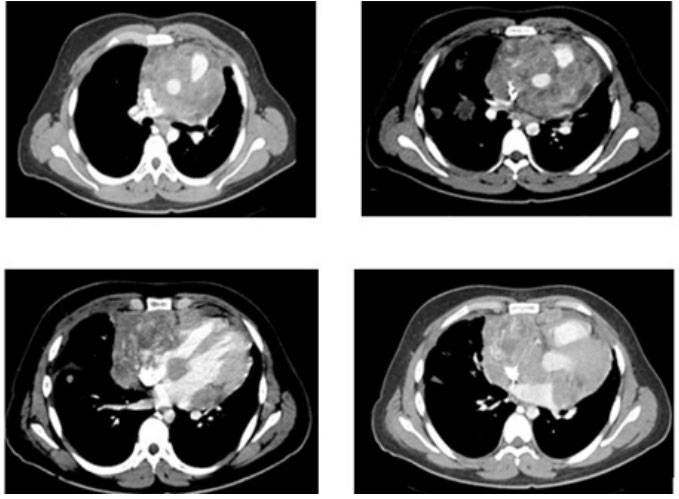 Figure 1: Contrast CT of the chest arterial phase axial with medistinal window
Figure 1: Contrast CT of the chest arterial phase axial with medistinal window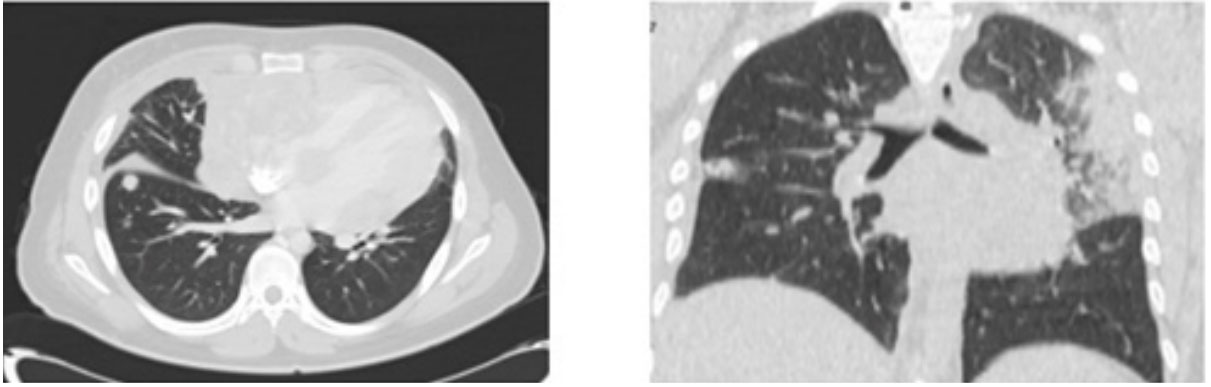 Figure 2: The Axial windows of the pulmonary parenchyma (A and B) show multiple spiculated nodules of central peripheral distribution and some with halos
of ground glass appearance suspicious of metastatic activity.
Figure 2: The Axial windows of the pulmonary parenchyma (A and B) show multiple spiculated nodules of central peripheral distribution and some with halos
of ground glass appearance suspicious of metastatic activity.A multidisciplinary evaluation of the case concluded the patient not to be a suitable candidate for initial surgical treatment but rather to treat with first-line chemotherapy and after response, surgical approach and consolidation radiotherapy would be entertained. A diagnostic thoracoscopy was performed with a pathology report of Epithelioid Metastatic Angiosarcoma with the following immunohistochemistry: CD34+, CD31+, Vimentin Gene +, ACE+, BER-EP4+, CD45+, p40-, PLAP-, FBHGC-, Ki67 50% (Figure 3-8).
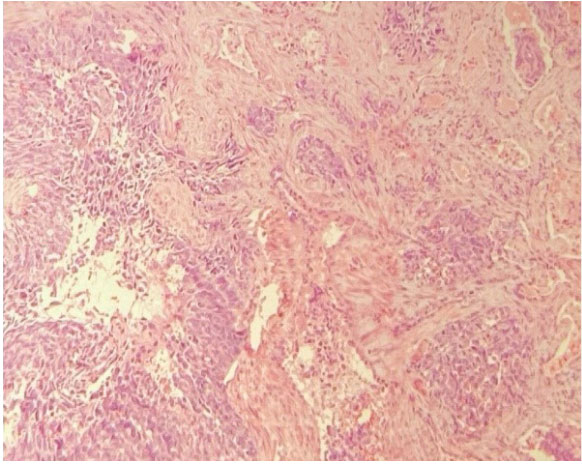 Figure 3: Malignant Cells of Vascular Growth Epitheliod Aspect.
Figure 3: Malignant Cells of Vascular Growth Epitheliod Aspect.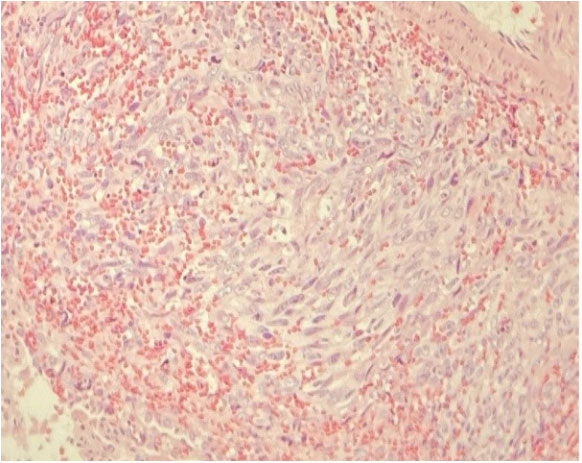 Figure 5: Malignant Vascular Neoformation Composed of Fisuform Cells.
Figure 5: Malignant Vascular Neoformation Composed of Fisuform Cells.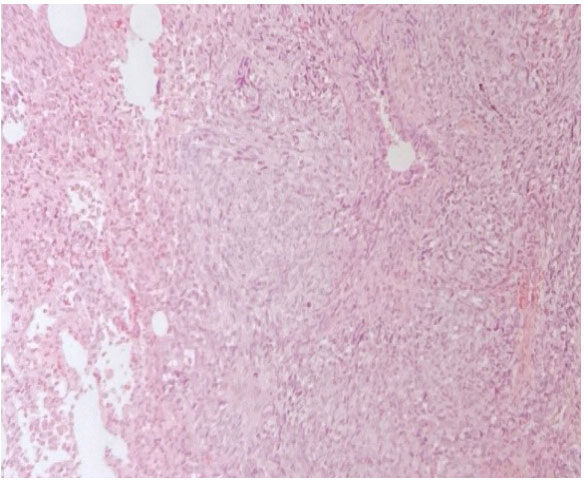 Figure 4: Peribronchial Infiltration of Vascular Neoplasia of Epithelioid
Aspect.
Figure 4: Peribronchial Infiltration of Vascular Neoplasia of Epithelioid
Aspect.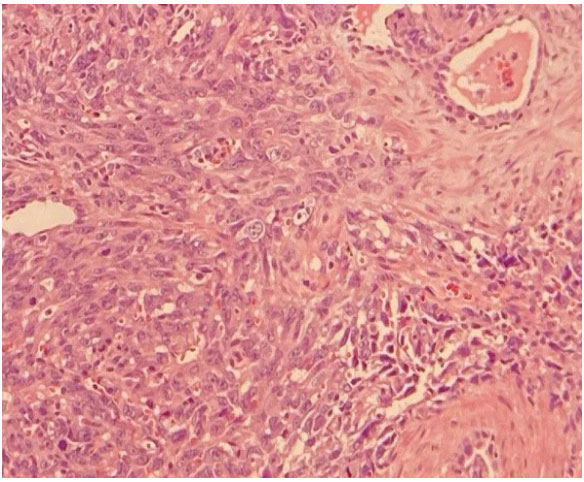 Figure 6:Malignant Epithelioid Cells with Atypical Pleomorphism
and Mitosis.
Figure 6:Malignant Epithelioid Cells with Atypical Pleomorphism
and Mitosis.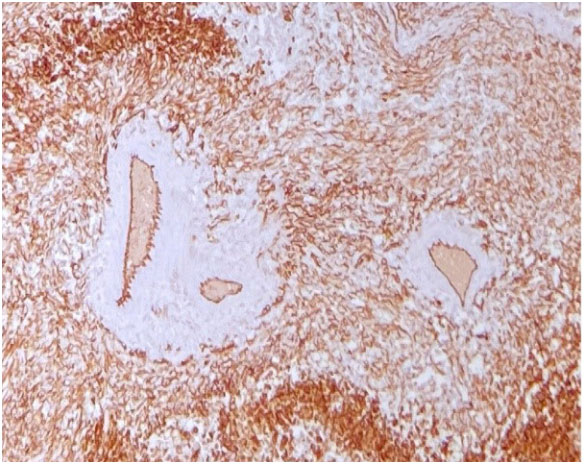 Figure 7: IHC CD31 Positive.
Figure 7: IHC CD31 Positive.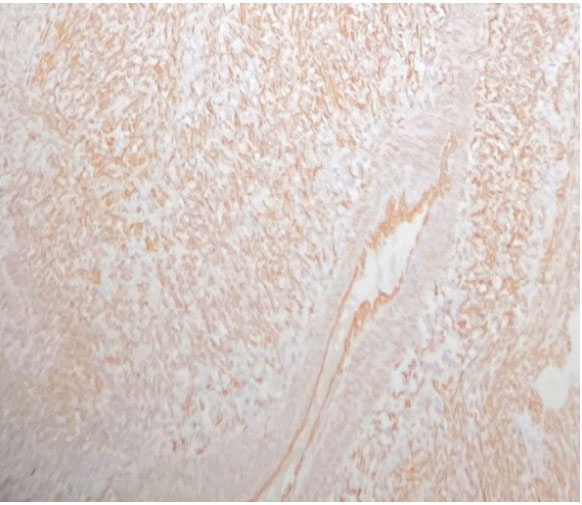 Figure 8: Vimentin Positive.
Figure 8: Vimentin Positive.The patient had an adequate functional status (ECOG 1) and first-line chemotherapy was initiated with 6 cycles of Epirubicin, Ifosfamide, and Cisplatin, which was well tolerated with the improvement of symptoms especially hemoptysis. Radiological evaluation showed stable disease and the patient was offered a second line of treatment based on Gemcitabine and Docetaxel but this was declined by the patient and only palliative therapy was instituted. The patient died 2 months after the conclusion of chemotherapy and had an overall survival of only 8 months.
Due to the limited number of cases of this disease, there are no current standard regimens specifically for primary pulmonary angiosarcoma. For locally confined disease, surgery has been the first approach in such patients, followed by radiotherapy, as angiosarcomas are radiosensitive. For advanced-stage disease,another than the combination of doxorubicin and ifosfamidebased regimens, there have been few other chemotherapeutic options for the treatment of local or metastatic angiosarcoma [2].
Angiosarcomas represent less than 1% of all sarcomas and develop most often in the skin, soft tissue, or liver. They may be associated with previous radiation treatment; environmental carcinogens like vinyl chloride, thorotrast, or phenylethyl hydrazine, foreign body material, or lymphedema.
The lungs are more often the site of metastasis from extrapulmonary tumors, most frequently from the heart and the pulmonary artery trunk. In the lung, primary angiosarcomas are extremely uncommon. The age of patients affected ranges between 22 to 79 years (mean, 54 years) with a male to female ratio of 3:1 [7]. Primary pulmonary angiosarcomas and metastatic angiosarcomas in the lung have similar symptomatology and radiologic features. The most common presentation clinically includes hemoptysis, shortness of breath, and weight loss [4].
Since the patient had a poor response to first-line therapy with Epirubicin, Ifosfamide, and Cisplatin, the tumor could not be resected as salvage therapy nor radiotherapy was used for consolidation or local control as initially planned. Unfortunately, the patient declined a second-line therapy with Gemcitabine/ Docetaxel that some reports have shown good response rates.

