 Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY
Journal of Medical Research and Surgery
PROVIDES A UNIQUE PLATFORM TO PUBLISH ORIGINAL RESEARCH AND REMODEL THE KNOWLEDGE IN THE AREA OF MEDICAL AND SURGERY



 Indexed Articles
Indexed ArticlesSelect your language of interest to view the total content in your interested language
Veronica Salais1*, Cesar A. Lopez2 , Carlos T. Perzabal2 1Division of Internal Medicine/Oncology, Hospital General de Ciudad Juarez, Avenida Paseo Triunfo de la Republica, 32340 Ciudad Juarez, Chihuahua, Mexico 2Division of General Surgery/Laparoscopy, Hospital General de Ciudad Juarez, Avenida Paseo Triunfo de la Republica, 32340 Ciudad Juarez, Chihuahua, Mexico
Author contributions: All authors contributed to the acquisition of data, writing, and revision of this manuscript.
Correspondence to: Veronica Salais, Internal Medicine Resident, Division of Internal Medicine/Oncology, Hospital General de Ciudad Juarez, Avenida Paseo Triunfo
de la Republica, 32340 Ciudad Juarez, Chihuahua, Mexico; E-mail: vero_bjs@msn.com
Received date: August 10, 2020; Accepted date: August 18, 2020; Published date: August 26, 2020
Citation: Salais V, Lopez CA, Perzabal CT (2020) Parathyroid Carcinoma. Clinical Image. J Med Res Surg 1(5): pp. 1-3.
Copyright: ©2020 Salais V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Parathyroid Carcinoma, Hyperparathyroidism, Pregnancy, Pancreatitis, Nephrocalcinosis
HPT-JT: Hyperparathyroidism-Jaw Tumor Syndrome; MEN1:Multiple Endocrine Neoplasia Type 1; MEN2A: Multiple Endocrine Neoplasia Type 2A; RB: Retinoblastoma; PRAD1: Cyclin Dl/Parathyroid Adenomatosis Gene 1; PTH: Parathyroid Hormone; HCG: Human Chorionic Gonadotropin; PT: Parathyroid.
Parathyroid carcinoma is an uncommon malignancy, less than 1000 recorded cases reported in the literature since described by de Quervain in 1904. With an estimated prevalence of 0.005% of all cancers, is the rarest endocrine cancer, and accounts for 0.5-5% of all circumstances of primary hyperparathyroidism [1].
The pathogenesis of parathyroid cancer is unknown, it may occur sporadically or as a part of a genetic syndrome. Genetic syndromes that have reported an association with parathyroid cancer cases include Hyperparathyroidism, Jaw Tumor Syndrome (HPT-JT), MEN1, MEN2A, and isolated familial hyperparathyroidism, parathyroid carcinoma occurs in 15% of HPT-JT patients with hyperparathyroidism (Figure 1).
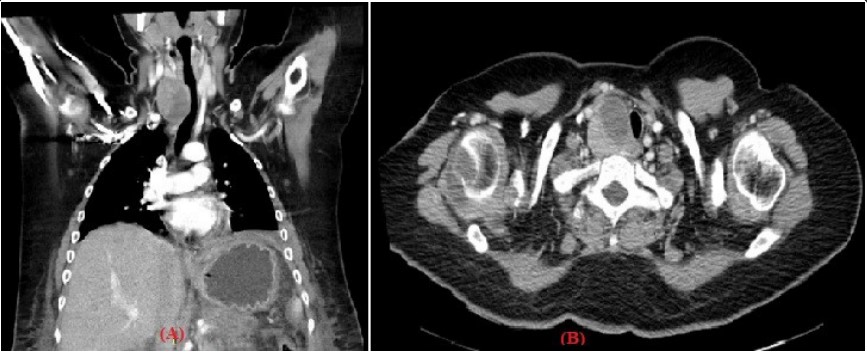 Figure 1: (A/B): Chest tomography in coronal reconstruction and axial section in arterial phase showing a solid infrahyoid mass in the visceral space at the level
of the right thyroid lobe extended to the retrotracheal space with homogeneous reinforcement to the application of contrast, which compresses and displaces
the airway to the left.
Figure 1: (A/B): Chest tomography in coronal reconstruction and axial section in arterial phase showing a solid infrahyoid mass in the visceral space at the level
of the right thyroid lobe extended to the retrotracheal space with homogeneous reinforcement to the application of contrast, which compresses and displaces
the airway to the left.Other than in genetic syndromes, no established risk factors have been identified thus far. However, cases of parathyroid cancer arising in patients with prior neck radiation exposure or secondary and tertiary hyperparathyroidism from chronic renal failure have been reported [2].
Oncogenes and tumor suppressor genes have been linked to parathyroid carcinomas, especially those involved in the control of the cell cycle. Examples include Retinoblastoma (RB), p53, Breast Carcinoma Susceptibility (BRCA2), and Cyclin Dl/ Parathyroid Adenomatosis Gene 1 (PRAD1) genes [3]
The hypercalcemic symptoms in cancer patients are often more pronounced than in those cases arising from benign parathyroid tumors. Parathyroid carcinoma patients tend to show simultaneous manifestations of renal and skeletal involvement at the time of presentation. Polyuria, renal colic, nephrocalcinosis, and nephrolithiasis are common renal complications. Bone pain, osteopenia, and pathologic fractures are manifestations of skeletal complications. Digestive complications include nausea, abdominal pain, peptic ulcers, and pancreatitis. Psychiatric
symptoms include fatigue and depression. Many parathyroid carcinoma patients initially present with a hypercalcemic crisis, also known as parathyrotoxicosis. Although most patients with benign parathyroid tumors have mild to asymptomatic disease, some may present with the same severe symptomatology of hypercalcemia with comparable metabolic manifestations [2] Non-functioning parathyroid carcinoma usually presents at a more advanced stage with symptoms of compression or invasion of adjacent structures, neck mass and/or dysphagia,hoarseness, or dyspnoea [1].
Parathyroid carcinoma is often difficult to diagnose preoperatively. The diagnosis is suggested by a combination of clinical and biochemical features, mainly markedly raised serum calcium levels (usually >14 mg/dl) and Parathyroid Hormone (PTH) [4]. The serum PTH level is also significantly elevated, commonly 3 to 10 times the upper limit of normal. Other abnormally elevated biochemical serum markers include alkaline phosphatase and α- and β- subunits of Human Chorionic Gonadotropin (HCG) [2].
The use of more than one imaging method increases diagnostic sensitivity and accuracy. Some ultrasound features may anticipate malignancy and help identify enlargement of lymph node or invasion into nearby structures: size >3 cm should raise the suspicion. Technetium-99 m-sestamibi (MIBI) allows localization and hence the distinction between eutopic and ectopic Parathyroid (PT) tissue, as well as identification of recurrent disease (Figure 2) [1].
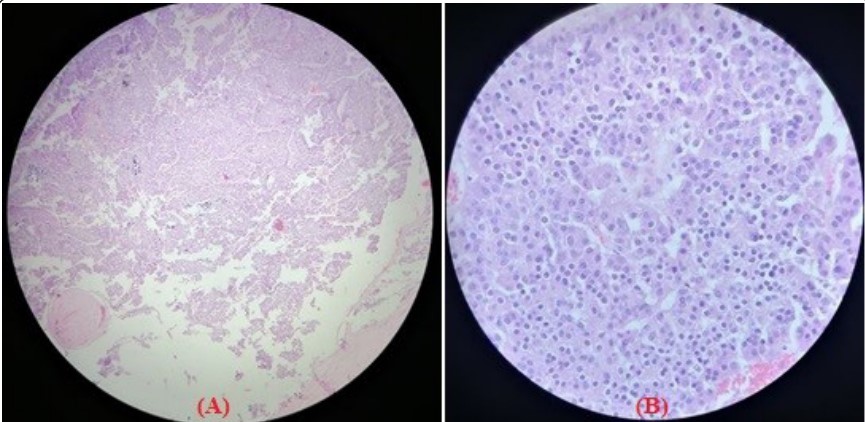 Figure 2: (A/B): Excisional parathyroid biopsy, conventional sections with hematoxylin and eosin staining, showing oncocytic cells with abundant cytoplasm,
nucleoli with pleomorphism, hyperchromatic nuclei, and prominent nucleolus.
Figure 2: (A/B): Excisional parathyroid biopsy, conventional sections with hematoxylin and eosin staining, showing oncocytic cells with abundant cytoplasm,
nucleoli with pleomorphism, hyperchromatic nuclei, and prominent nucleolus.Surgery is the only curative treatment for parathyroid carcinoma and consists of complete resection of the primary lesion at the time of initial operation. Patients who present with features suggestive of parathyroid carcinoma warrant thorough exploration of all four parathyroid glands, because parathyroid carcinoma has been reported to coexist along with benign adenomas or hyperplasia. The most effective surgical approach is bloc resection, tracheoesophageal, paratracheal, and upper mediastinal lymph nodes should be excised, but an extensive lateral neck dissection is indicated only when there is spread to the anterior cervical nodes. When the diagnosis of parathyroid cancer is made after parathyroid surgery based on pathology, as often happens, the management plan becomes more complex [3].
Despite a potentially curative resection, parathyroid carcinoma has a recurrence rate of >50%. Most recurrences occur 2-3 years after the initial operation, but this period is variable, and a prolonged disease-free interval of as long as 20 years has been reported [3].
Chemotherapy generally is disappointing and radiotherapy has little, if any, effect in invasive parathyroid cancer [3]. The prognosis of PC is variable. Overall survival rates are generally better than in most solid tumors, with 76 to 85% and 49 to 77% patients alive respectively at 5-year and 10-year follow-up [1]. The best prognosis depends on early recognition and complete excision of the tumor at initial surgery [3].
We present a 27-year-old female with a history of antiphospholipid syndrome in treatment, in her mediate cesarean postpartum due to preeclampsia who started her condition with dysphagia and dysphonia, as well as a weight loss of 15 kg in 5 months, neck tumor of 3 cm, asthenia adynamia, presenting laboratories upon admission compatible with acute kidney injury, pancreatitis and hyperparathyroidism.
Laboratories with leukocytosis 18.26, Neutrophils 16.71, Lymphocytes 0.81, Hemoglobin 9.90, Platelets 208, Glucose 82, Creatinine 2.06, urea nitrogen 25, Urea 54, Chlorine 116, Potassium 2.7, Sodium 145, Calcium 15.9, Phosphorus 5.8, Magnesium 2 (Figure 1). It is assessed by the surgery service who determines is a candidate for traditional cervical parathyroidectomy. CT image with bilateral pleural effusion of a higher degree on the left kidney stone with staghorn stone, right kidney with more severe calcinosis, pyelocalyceal ectasia due to possible urethral stenosis, acute pancreatitis grade D Baltazar.

