 Journal of Clinical and Biomedical Investigation
PROVIDES A UNIQUE PLATFORM COVERING SCIENTIFIC KNOWLEDGE IN BIOMEDICAL SCIENCES AND CLINICAL RESEARCH
Journal of Clinical and Biomedical Investigation
PROVIDES A UNIQUE PLATFORM COVERING SCIENTIFIC KNOWLEDGE IN BIOMEDICAL SCIENCES AND CLINICAL RESEARCH
Journal of Clinical and Biomedical Investigation
PROVIDES A UNIQUE PLATFORM COVERING SCIENTIFIC KNOWLEDGE IN BIOMEDICAL SCIENCES AND CLINICAL RESEARCH
Journal of Clinical and Biomedical Investigation
PROVIDES A UNIQUE PLATFORM COVERING SCIENTIFIC KNOWLEDGE IN BIOMEDICAL SCIENCES AND CLINICAL RESEARCH
Anubha Bajaj*
Consultant Histopathologist, AB Diagnostics, India.
Correspondence to: Anubha Bajaj, Consultant Histopathologist, AB Diagnostics, India.
Received date: May 16, 2022; Accepted date: May 29, 2022; Published date: June 6, 2022
Citation: Bajaj A (2022) The Sophomoric Neurons-Astroblastoma: A Mini Review. J Clin Biomed Invest 2(1): pp. 27-31. doi: 10.52916/jcbi224015
Copyright: ©2022 Bajaj A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use,
distribution and reproduction in any medium, provided the original author and source are credited.
Astroblastoma is an uncommon, controversial neoplasm of the Central Nervous System (CNS) emerging from the glia. “Astroblastoma” as a terminology was initially coined in 1924 for a tumefaction characteristically emerging as a unique astrocytic glioma comprised of tumour cells configuring perivascular pseudo-rosettes and appearing immune reactive to Glial Fibrillary Acidic Protein (GFAP). Bucy and Bailey in 1930 delineated diverse macroscopic and microscopic features of the neoplasm with description of individual astroblasts as unipolar cells with broad “feet” amalgamating adjacent to vascular articulations. Subsequently in 1933, Cox categorized astroblastoma as a neoplasm transitioning between astrocytoma and glioblastoma multiforme.
Astroblastoma, Tumour cells, Corpus callosum.
Astroblastoma is a distinct clinico-pathological entity associated with well exemplified radiographic, histopathological and cytogenetic features. Astroblastoma exhibits diverse histological manifestations wherein the biological behaviour pertains to an indolent or aggressive, anaplastic and malignant neoplasm. Nevertheless, tumour histogenesis, discernment, classification and cogent therapeutic strategies remain debatable. World Health Organization (WHO) categorizes astroblastoma as a neuroglial tumour wherein contemporary nomenclature of circumscribed astrocytic glioma or MN1-altered astroblastoma is adopted for classifying central nervous system tumours. Chromosomal gain of +20q and +19 is consistently observed [1,2].
Generally, tumefaction is amenable to cogent surgical resection. Malignant metamorphosis is occasional with the expression of pertinent histological features. Astroblastoma remains an exceptional, controversial neoplasm with variable clinical outcomes and uncertain cellular origin. Disease Characteristics Typically, astroblastoma is confined to cerebral hemispheres although tumefaction may occur within cerebellum, brainstem, corpus callosum, hypothalamus or ventricular system. Classically, supra-tentorial compartment of cerebral hemispheres is predominantly implicated. Nevertheless, tumour infiltration into corpus callosum, cerebellum, pineal gland, brain stem or ventricular system may ensue. Lobar tumefaction specifically emerges within frontal lobe and parietal lobe, followed by temporal lobe [3,4]. Children, adolescents and young adults are commonly incriminated. No age of tumour emergence is exempt and neoplasms are discerned from early childhood to sixth decade. Mean age of tumour discernment is up to 30 years whereas median age is 11 years. A female predominance is observed.
Of obscure cytogenesis, it is posited that astroblastoma is engendered from embryonic cells prospectively differentiating into astrocytes. Especially, embryonic cells comprising of an intermediate stage between unipolar spongioblasts and astrocytes may generate the neoplasm. Contingent to ultrastructural similarities between tanycytes and astroblasts, a cellular origin is indicated wherein the neoplasm is probably derived from tanycytes [3,4].
Of debatable origin, astroblastoma is contemplated to demonstrate an astrocytic or ependymal lineage. Astroblastoma is accompanied by diverse molecular characteristics and morphological concurrence with pleomorphic xanthoastrocytoma, ependymoma or diffuse astrocytoma. Frequent genomic anomalies such as chromosomal gain in 20q and 19 and deletions in chromosome 10 and X are observed. Also, loss of heterozygosity in 9p may emerge as a predictor to malignant metamorphosis. Astroblastoma exhibits distinct genetic subtypes, contingent to pV600E mutation within B-Raf serine/threonine kinase gene (BRAFV600E) or rearrangement of meningioma 1 (MN1) gene which is disrupted with balanced translocation. Occurrence of contemporary MN1 genomic alteration, a transcriptional co-regulator implicated in emergence of acute myeloid leukaemia or meningioma is observed. Genomic fusion between MN1 at chromosome region 22q12.1 and BEN domain containing 2 (BEND2) at Xp22.13 is encountered in MN1-rearranged neoplasms [3,4].
Clinical course of astroblastoma is non concordant to histological appearance. The neoplasm depicts an admixture of clinical features of an astrocytoma and ependymoma. Individuals exemplify headache with increasing intensity, nausea, vomiting, motor aphasia, dysnomia, hemi-hypoesthesia, pertinent corticospinal signs and hemiparesis preceded by tonic-clonic seizures.
Cardinal clinical symptoms appear as an amalgamation of headache, focal neurologic deficit and seizures. Significant mass effect may ensue within supra-tentorial compartment. Gradual tumour evolution ensures discernment in up to 18 months following initial, cogent clinical symptoms, thereby majority of detected neoplasms demonstrate mammoth dimensions.
Grossly, astroblastoma emerges as a well circumscribed, soft to firm, cystic, lobulated tumefaction with focal necrosis and haemorrhage. Generally, the solid, soft, greyish/white to pearly neoplasm exhibits distinctive haemorrhagic areas.
Focal cystic degeneration, haemorrhage and necrosis is observed within well demarcated, “bubbly” tumefaction. Tumour infiltration within encompassing cerebral hemispheres or adjacent brain is exceptional (Figure 1,2).
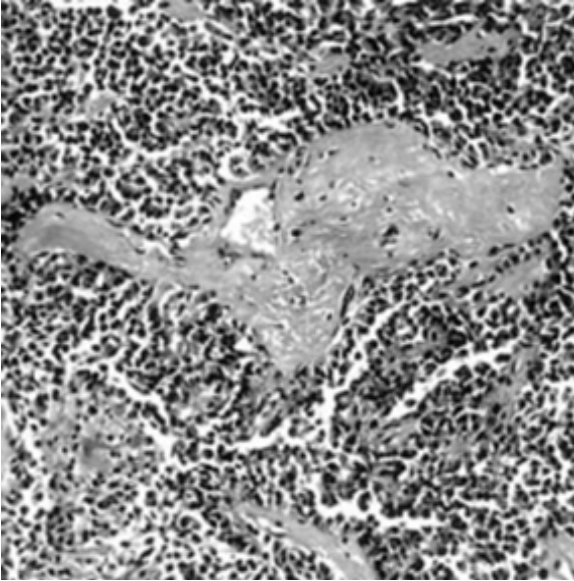 Figure 1: Astroblastoma exhibiting perivascular pseudo-rosettes composed of elongated glial cells and broad, cytoplasmic processes radially extending towards centric vascular articulations.
Figure 1: Astroblastoma exhibiting perivascular pseudo-rosettes composed of elongated glial cells and broad, cytoplasmic processes radially extending towards centric vascular articulations.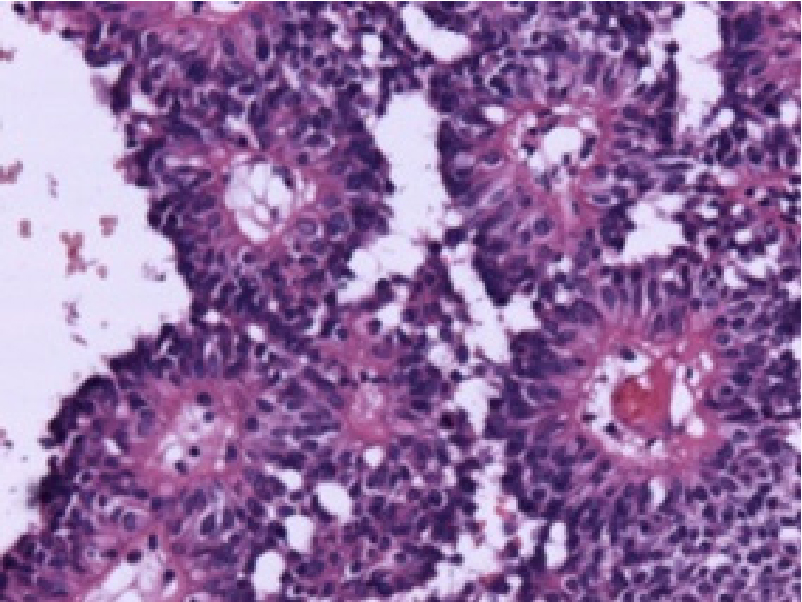 Figure 2: Astroblastoma displaying perivascular pseudo-rosettes comprised of elongated glial cells with centric vascular configurations
Figure 2: Astroblastoma displaying perivascular pseudo-rosettes comprised of elongated glial cells with centric vascular configurationsUpon microscopy, hyper-vascular, papillary neoplasm is composed of minimally pleomorphic cells imbued with pleomorphic, spherical to elliptical nuclei and prominent nucleoli. Atypical mitotic figures may appear. Perivascular pseudorosettes may be significant. Endothelial cell hyperplasia and hyalinisation of vascular articulations is prominent (Figure 3,4).
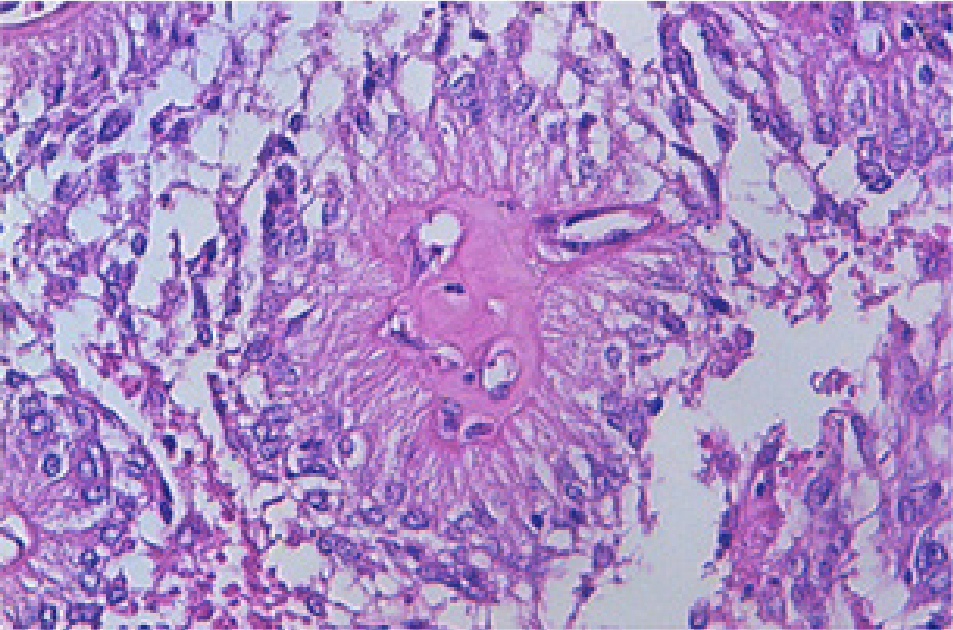 Figure 3: Astroblastoma depicting centroidal, hyalinised vascular articulations with peripherally disseminated elongated glial cells with abundant eosinophilic cytoplasm.
Figure 3: Astroblastoma depicting centroidal, hyalinised vascular articulations with peripherally disseminated elongated glial cells with abundant eosinophilic cytoplasm.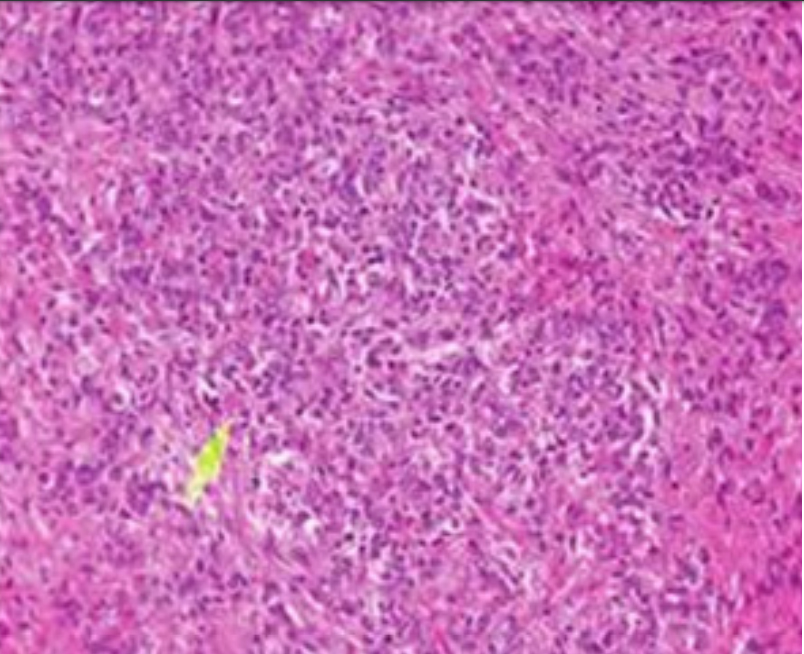 Figure 4: Astroblastoma delineating clusters of spindle-shaped glial cells
with circumscribing thick walled vascular articulations with endothelial
prominence
Figure 4: Astroblastoma delineating clusters of spindle-shaped glial cells
with circumscribing thick walled vascular articulations with endothelial
prominenceCharacteristically, tumour cells circumscribe a centric capillary and configure petal-like structure denominated as the ‘astroblastic pseudo-rosette’ . The neoplasm demonstrates a papillary pattern with prominent collagen deposition and intramural hyalinization. Microscopic attributes such as perivascular pseudo-rosettes, perivascular hyalinization and lack of stromal fibrillary metamorphosis appear significant for appropriate tumour categorization (Figure 5,6).
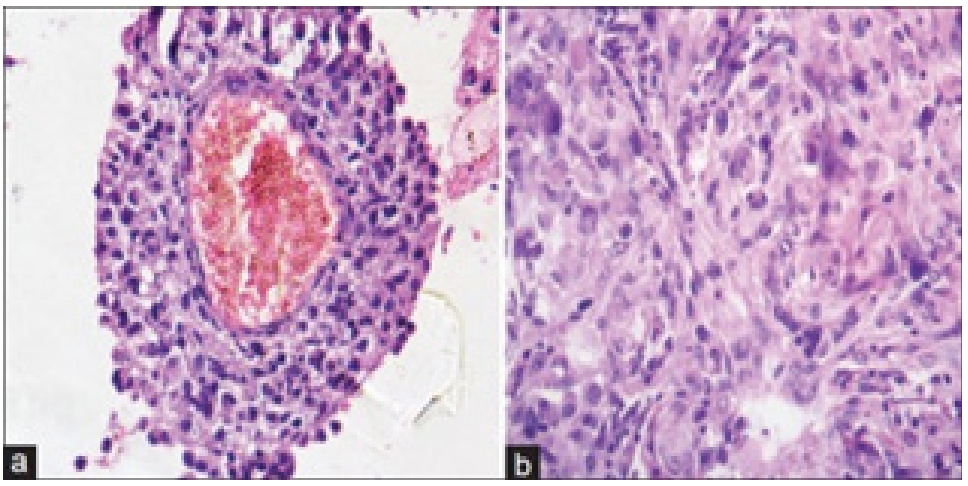 Figure 5: Astroblastoma demonstrating perivascular pseudo-rosettes comprised of spindle-shaped glial cells with abundant, eosinophilic cytoplasm.
Figure 5: Astroblastoma demonstrating perivascular pseudo-rosettes comprised of spindle-shaped glial cells with abundant, eosinophilic cytoplasm. Figure 6: Astroblastoma exemplifying perivascular pseudo-rosettes constituted of elongated glial cells which are immune reactive to glial fibrillary
acidic protein
Figure 6: Astroblastoma exemplifying perivascular pseudo-rosettes constituted of elongated glial cells which are immune reactive to glial fibrillary
acidic proteinGenerally, astroblastic cells are polarized, poorly cohesive, devoid of epithelial surface differentiation and monopolar with singular cytoplasmic process which adheres to vascular articulations. Pseudo-rosettes are comprised of glial cells which configure a corona circumscribing capillaries layered with flattened endothelial cells superimposed upon thickened basal membrane.
Astroblastoma is composed of elongated glial cells with abundant eosinophilic cytoplasm with broad, “stout”, cytoplasmic processes which extend radially towards a centroidal vascular articulation with the configuration of characteristic "astroblastic pseudo-rosette", simulating perivascular pseudo-rosettes encountered in ependymoma. Centric vascular articulation embedded within cellular pseudo-rosettes appears hyalinised or sclerotic.
The well circumscribed neoplasm with discrete, pushing perimeter preponderantly exhibits perivascular pseudorosettes with thick cellular processes extending from cell body towards vascular tunica adventitia. Focal vascular hyalinization or minimal fibrillary substances are delineated (Figure 7,8).
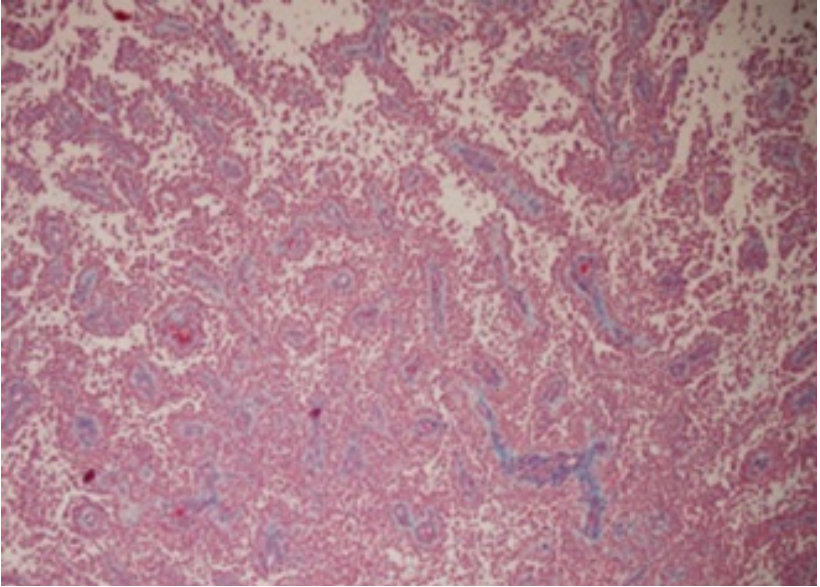 Figure 7: Astroblastoma enunciating elongated cords and clusters of glial cells with cytoplasmic processes, abundant eosinophilic cytoplasm and a perivascular amalgamation.
Figure 7: Astroblastoma enunciating elongated cords and clusters of glial cells with cytoplasmic processes, abundant eosinophilic cytoplasm and a perivascular amalgamation. Figure 8: Astroblastoma exhibiting delicate cords and clusters of monopolar, poorly cohesive spindle-shaped glial cells surrounding hyalinised vascular articulations
Figure 8: Astroblastoma exhibiting delicate cords and clusters of monopolar, poorly cohesive spindle-shaped glial cells surrounding hyalinised vascular articulationsContingent to histologic characteristics, astroblastoma is subdivided into:
High grade astroblastoma is hyper-cellular with frequent mitotic activity, focal vascular proliferation or tumour necrosis with pseudo-palisading of cells. Signet ring cells are exceptional. Occasional foci of tumour infiltration are observed in high grade lesions.
Tumour reoccurrence is associated with possible transformation into anaplastic, high grade variant of astroblastoma.
Upon ultrastructural examination, tumour cells exhibit an irregular cytoplasm, prominent nucleolus, cytoplasmic interdigitations upon lateral cellular perimeter and inconstant, inadequate intercellular junctions. Abundant intermediate filaments configure fascicles confined to tumour cell cytoplasm. Membrane junctions and external lamina of tumour cells appear adherent to collagen fibres [5,6].
Tumour cells are mildly, focally immune reactive to Epithelial Membrane Antigen (EMA), S100 protein and diffusely immune reactive to vimentin. Tumour cells express a non-fibrillary variant of Glial Fibrillary Acidic Protein (GFAP). Thus, epithelioid cells are diffusely immune reactive to Glial Fibrillary Acid Protein (GFAP), especially within perivascular zones. Tumour cells are immune non reactive to synaptophysin, cytokeratin and Phospho-Tungstic Acid Haematoxylin (PTAH). Ki-67 labelling index of around ~40% can be enunciated [7,8].
Upon imaging, astroblastoma requires segregation from central nervous system neoplasms as primitive neuroectodermal tumour, supra-tentorial ependymoma, atypical teratoid/ rhabdoid tumour, gemistocytic astrocytoma, glioblastoma multiforme, oligodendroglioma with nodular calcification or pleomorphic xanthoastrocytoma [9,10].
Demarcation is required from:
Predominantly, tumefaction appears as an admixture of cystic and solid components followed in frequency by singularly solid tumefaction or cystic neoplasms. Tumefaction may represent an initial haemorrhagic appearance [11,12]. Imaging of cerebral lobes demonstrates the neoplasm in a majority of subjects. Frequently, peripheral lesions arising adjacent to cerebral convexity appear nodular, vascularized and well demarcated with tumour progression. Upon radiographic assay, a characteristic supra-tentorial, multi-lobulated lesion constituted of solid and cystic components is exemplified. Astroblastoma is a peripheral, superficial, supra-tentorial, lobulated, solid to cystic neoplasm of variable magnitude with minimal associated vasogenic oedema. Commonly discerned multiple cysts engender a “bubbly” appearance. Punctate calcification is enunciated in a majority (85%) of instances [10,11].
Upon Computerized Tomography (CT), a supra-tentorial, multilobar, well demarcated neoplasm with punctate calcification is comprised of solid and cystic components and appears confined to frontal or parietal region. The cystic component demonstrates mixed densities whereas solid component is hyper-dense as compared to white matter. Minimal peritumoral oedema is observed. Midline shift may occur along with collapse of ipsilateral ventricular system. Magnetic Resonance Imaging (MRI) delineates a well defined, intra-axial neoplasm of variable magnitude. Upon T1 weighted imaging, the heterogeneous neoplasm is predominantly hypo-intense to isointense to white matter. Upon T2 weighted imaging, a multi-cystic, hyper-intense to isointense lesion with prominent “bubbly”, heterogeneous tumour pattern is discerned [10,11]. Fluid Attenuated Inversion Recovery (FLAIR) sequences appear isointense within the lesion with minimal peritumoral oedema. Imaging with gadolinium contrast administration delineates a solid tumefaction with peripheral rim enhancement of the cystic component. Spectroscopy demonstrates an elevated peak with choline [11,12].
Comprehensive surgical resection is an optimal and recommended treatment strategy as the expansive, non infiltrative neoplasm depicts a well demarcated arachnoid plane upon imaging. Clinical symptoms such as hemiparesis and altered language may ameliorate following surgical intervention. Adjuvant therapy appears efficacious in lesions of advanced grade. Adjuvant radiation therapy or chemotherapy can be suitably adopted for treating high grade tumefaction. Nevertheless, adoption of chemotherapy remains non annotated. Tumour reoccurrence occurs in nearly 30% neoplasms and is generally observed in anaplastic tumours. Notwithstanding, tumour reoccurrence of low grade neoplasms following comprehensive surgical eradication is uncommon [11,12].

